https://quibio.web.uah.es/group/
y actualice sus enlaces.
Publicaciones > García-Marín et al
Pyrrolo[1,2-a]quinoxalines: Insulin Mimetics that Exhibit Potent and Selective Inhibition against Protein Tyrosine Phosphatase 1B.
1. Departamento de Química Orgánica y Química Inorgánica, Universidad de Alcalá, 28805, Alcalá de Henares, Spain. 2. Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), Ctra. Colmenar Viejo, km. 9100, 28034, Madrid, Spain. 3. Instituto de Investigación Química Andrés M. del Río, Facultad de Farmacia, Universidad de Alcalá, 28805, Alcalá de Henares, Spain. 4. Departamento de Biología de Sistemas, Universidad de Alcalá, 28805, Alcalá de Henares, Spain. 5. Departamento de Química y Bioquímica, Facultad de Farmacia, Universidad San Pablo CEU, 28925, Alcorcón, Spain. 6. Departamento de Química Analítica, Química Física e Ingeniería Química, Universidad de Alcalá, 28805, Alcalá de Henares, Spain.
Keywords: PTP1B; biological activity; inhibitors; insulin mimetics; pyrrolo[1,2-a]quinoxaline
Abstract
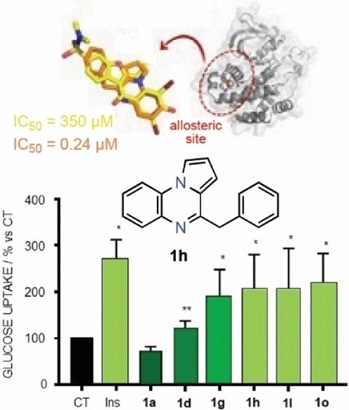
PTP1B dephosphorylates insulin receptor and substrates to modulate glucose metabolism. This enzyme is a validated therapeutic target for type 2 diabetes, but no current drug candidates have completed clinical trials. Pyrrolo[1,2-a]quinoxalines substituted at positions C1-C4 and/or C7-C8 were found to be nontoxic to cells and good inhibitors in the low- to sub-micromolar range, with the 4-benzyl derivative being the most potent inhibitor (0.24 μm). Some analogues bearing chlorine atoms at C7 and/or C8 kept potency and showed good selectivity compared to TCPTP (selectivity index >40). The most potent inhibitors behaved as insulin mimetics by increasing glucose uptake. The 4-benzyl derivative inhibited insulin receptor substrate 1 and AKT phosphorylation. Molecular docking and molecular dynamics simulations supported a putative binding mode for these compounds to the allosteric α3/α6/α7 pocket, but inconsistent results in enzyme inhibition kinetics were obtained due to the high tendency of these inhibitors to form stable aggregates. Computational calculations supported the druggability of inhibitors.